Usage
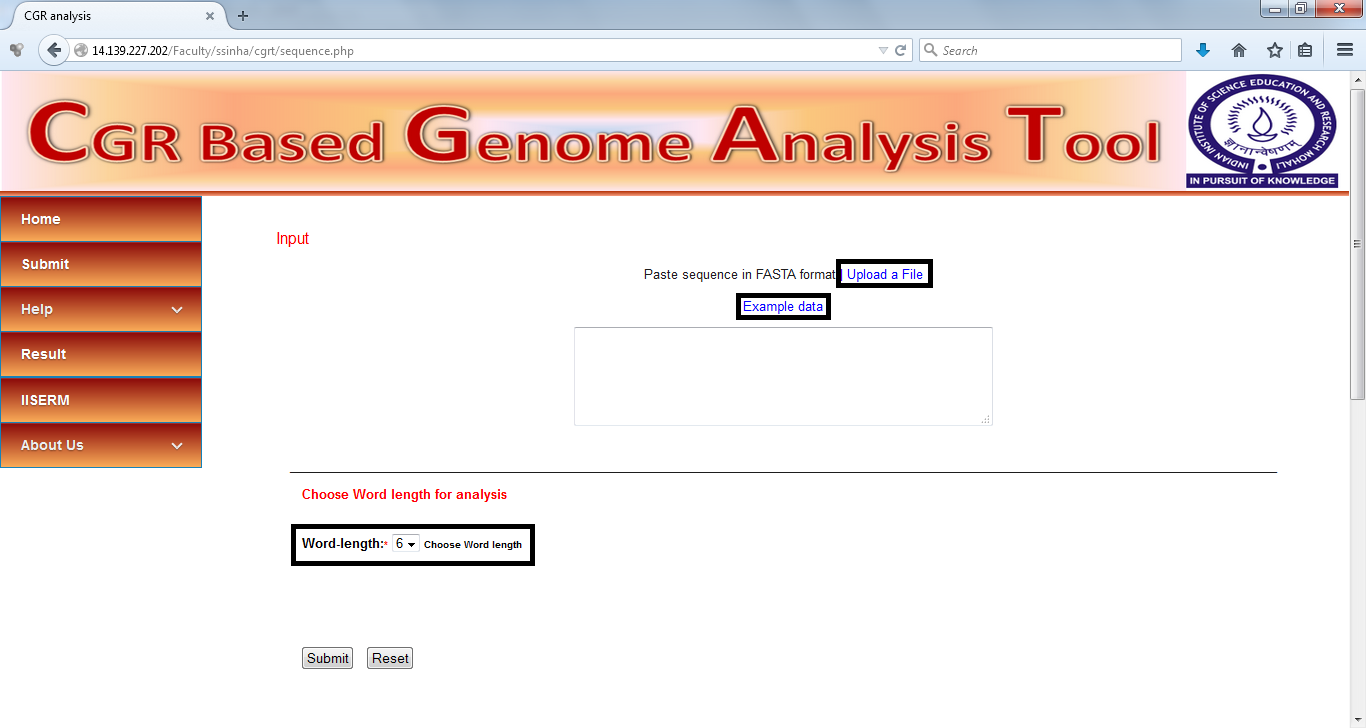
Input
The DNA sequences to be analyzed can be either pasted in the text box on "Submit" page or uploaded in the form text file. All the sequences must be in FASTA format. The allowed DNA characters in file are A, T, G, C, R, Y and N.Whole genome sequences of HIV-1 subtypes A, B, C, D, F, G, SIV and HTLV (Dataset used in Pandit et al Mol. Phylo. Evol., 2012) have been provided as an example. Click on the "Example data" link on Submit page to upload this data for analysis.
Selection of "Word Length"
The clustering of the sequences is based on the distances calculated from the frequencies of DNA words. The word length to be used for the calculation can be specified by the user at the submission page. This default word length used is 6. After selecting the word length click on "Submit"
Selection of Out group
An out group needs to be specified for construction of Neighbor Joining Tree by Phylip. Select the out group from the dropdown menu and confirm the submission. The calculation is started immediately and each submission is provided a unique Job ID.
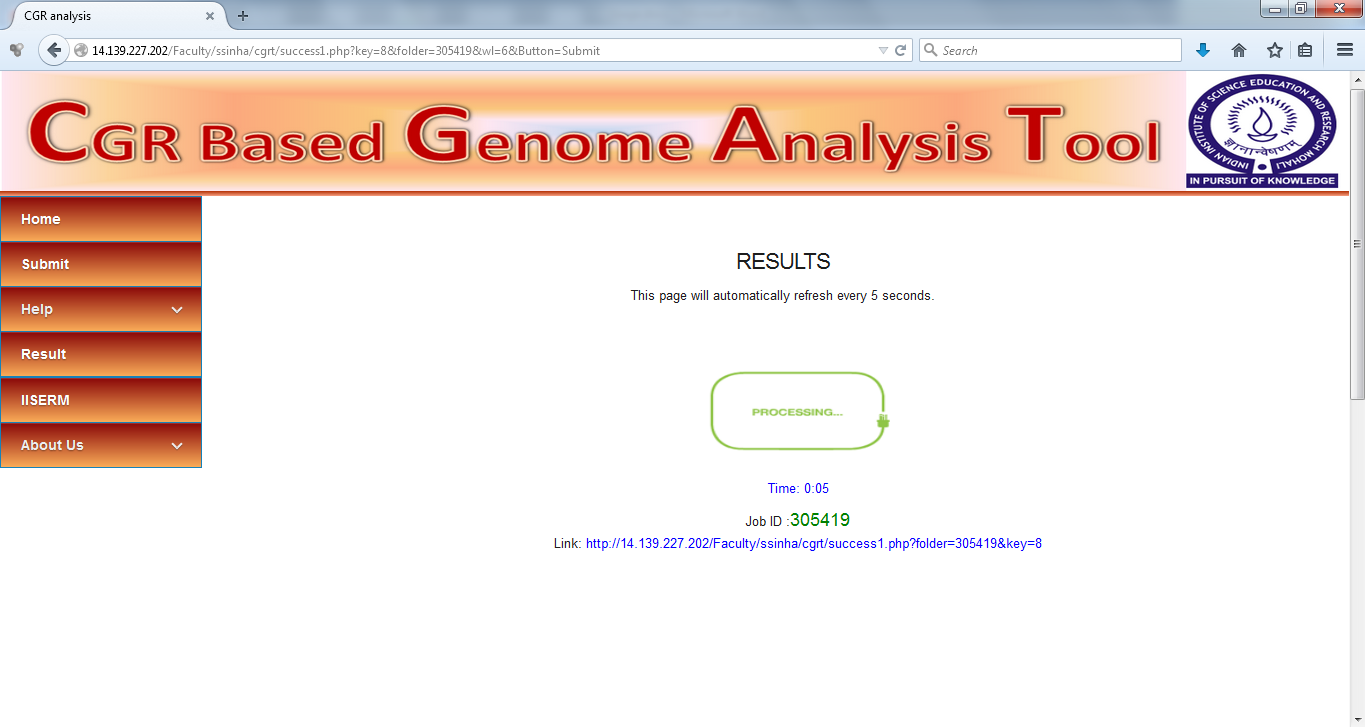
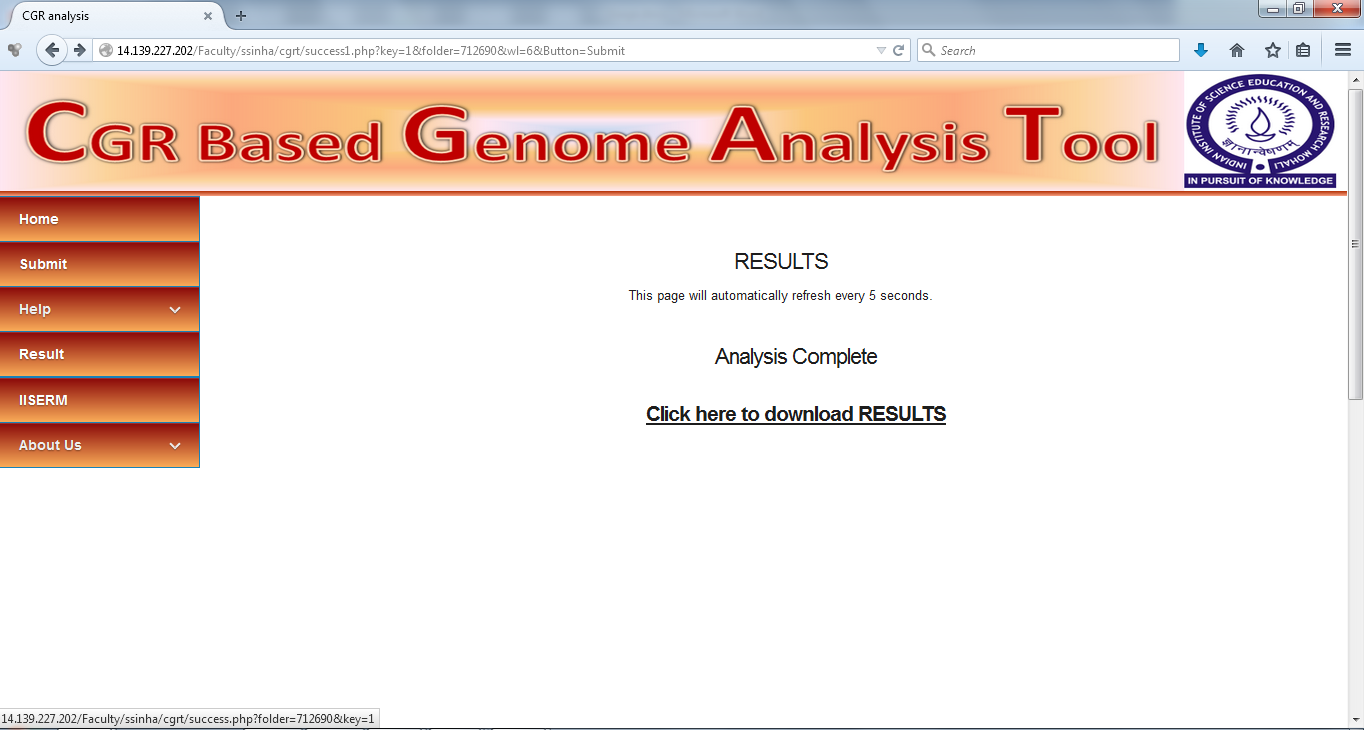
Results
Once the Analysis is complete the results page shows a link to all the results. Click on the link to access results.
The results comprise of following
Visualization of CGR plot
CGRs for each sequence can be visualized by selecting the sequence from dropdown menu.
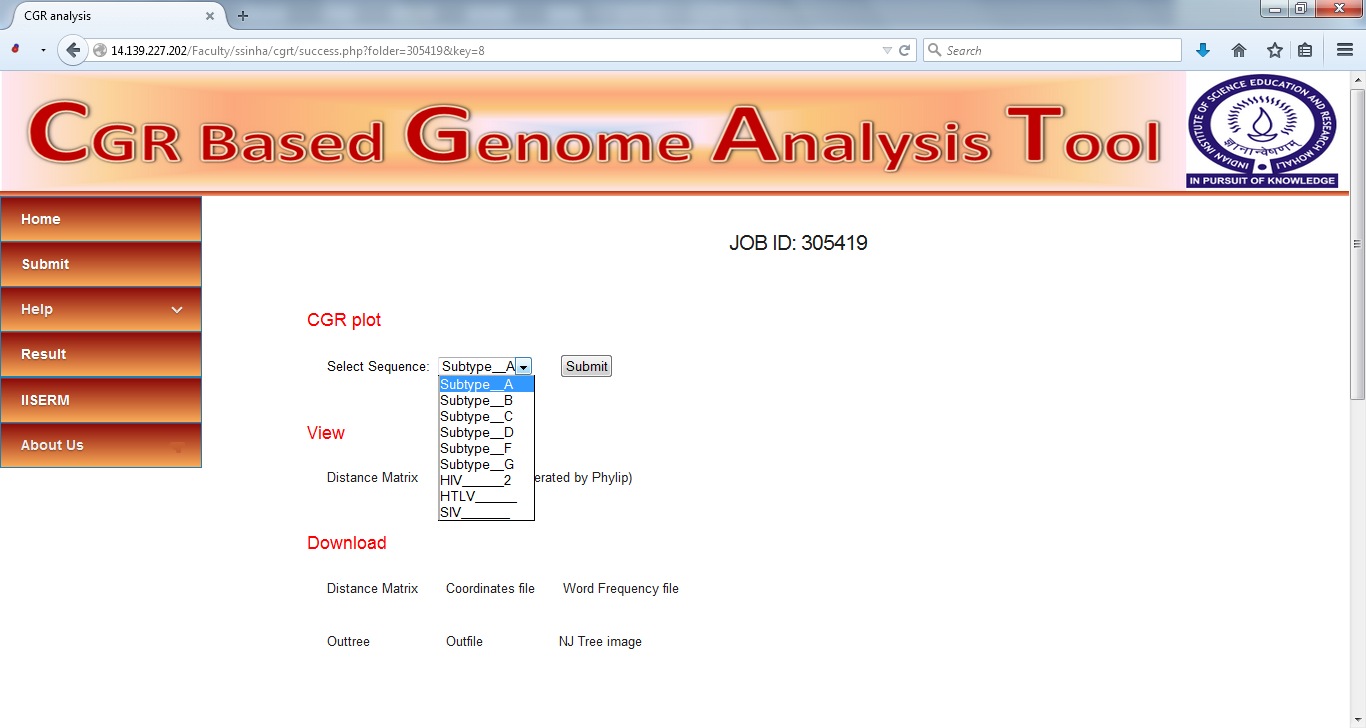
CGR plot for the Whole genome sequence of HIV-1 Subtype A as visualized on the browser, has been shown in the following screenshot
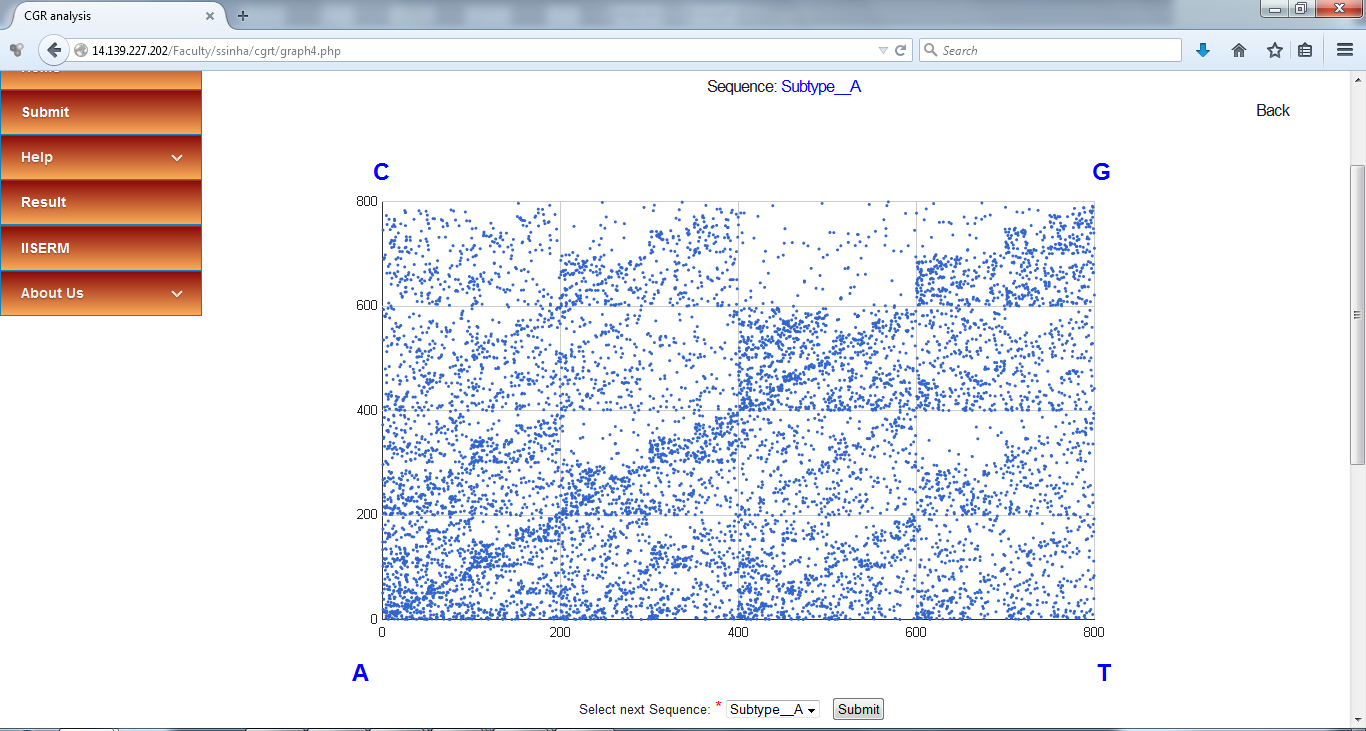
This plot can be saved by right click and selecting "Save image as" option.
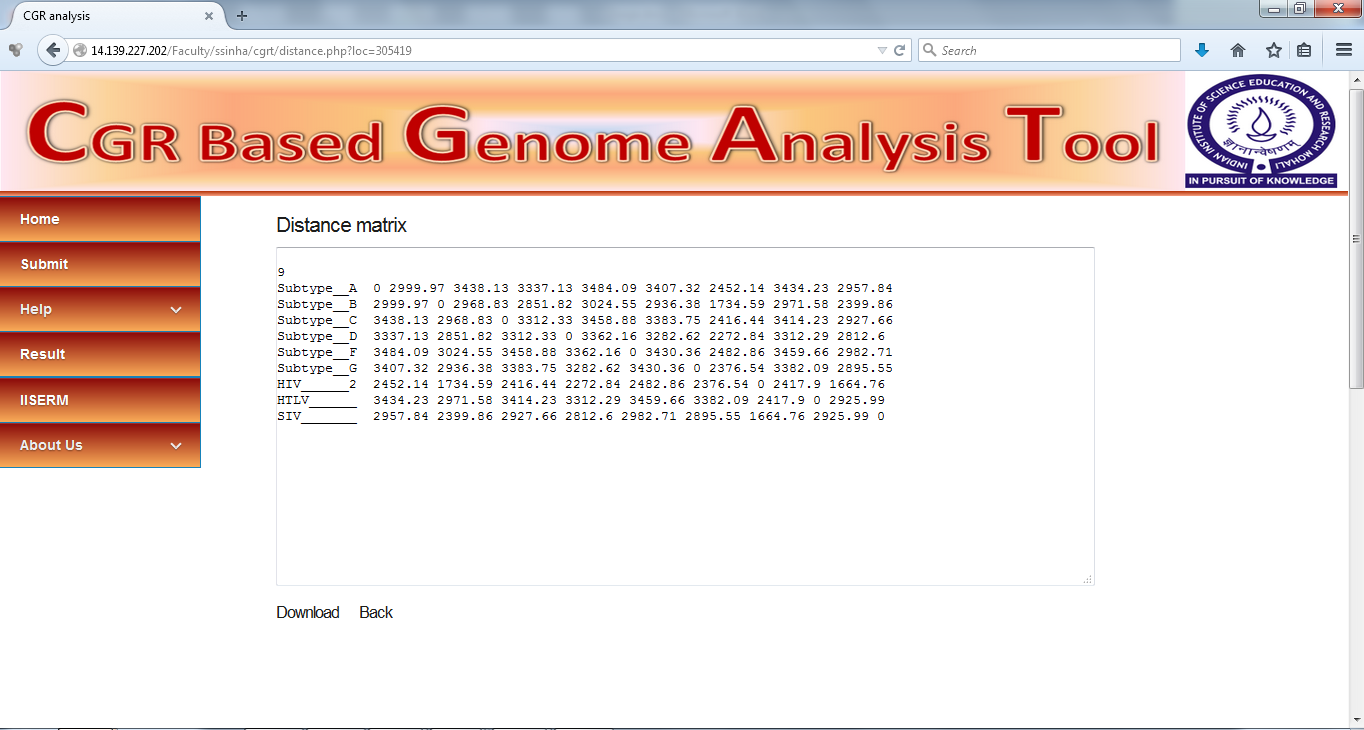
View Distance Matrix
Distance matrix generated by the CGR method can be used directly into phylip can be viewed on the browser. It can also be downloaded directly from this page.
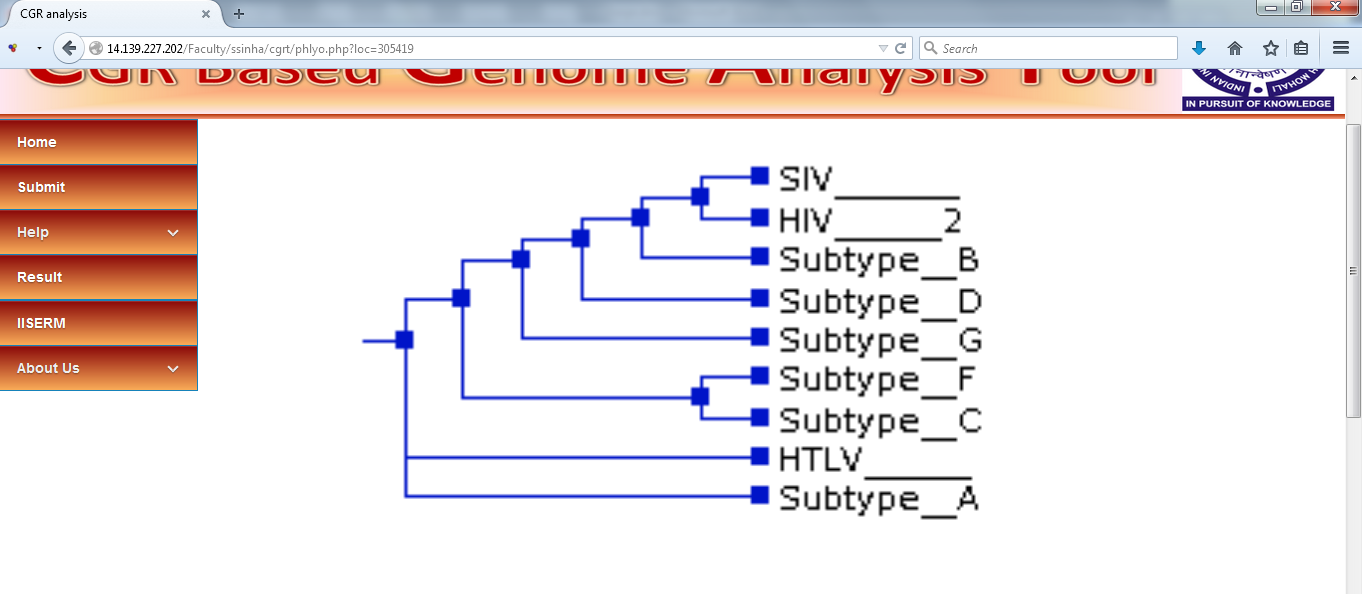
View NJ Tree
NJ tree, created by Phylip using the distance matrix shown above, can be visualized on the browser. This visualization is created using the Notung (version 2.6) package. This image can also be downloaded directly from this page in PNG format.
Download Results
All the results can be downloaded from the same page. The file that can be downloaded are
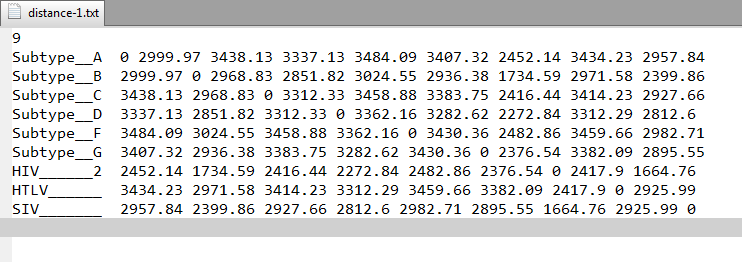
1) Distance Matrix file- It is (.txt) text file that contains pairwise distance matrix generated using input sequences. We used Euclidean distance method to calculate pairwise distances between multiple whole genome sequences. It contains output in standard PHYLIP distance matrix format. Following is the screenshot of the file for example data.
2) Coordinates file- Coordinate file is a (.txt) text file that contains x,y coordinates for each point in CGR of each sequence. It contains co-ordinates in following format.
>First Sequence name
X-coordinates, Y-coordinates
...
...
>Second Sequence name
X-coordinates, Y-coordinates
...
...
3) Word Frequency file- It is a (.txt) text file that contains frequencies of all different k-letter words corresponding to CGR map.
For example – At k=3 in example data set
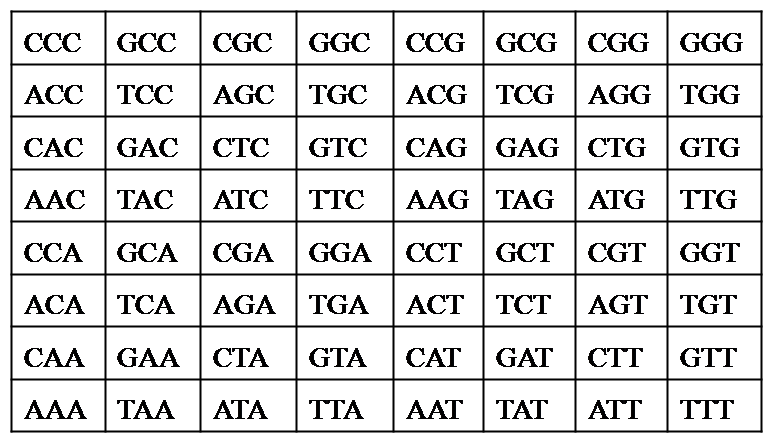
(2-d matrix of all possible 3 letter words using nucleotides A,T,G,C)
For the example data at word length 3
Subtype__A
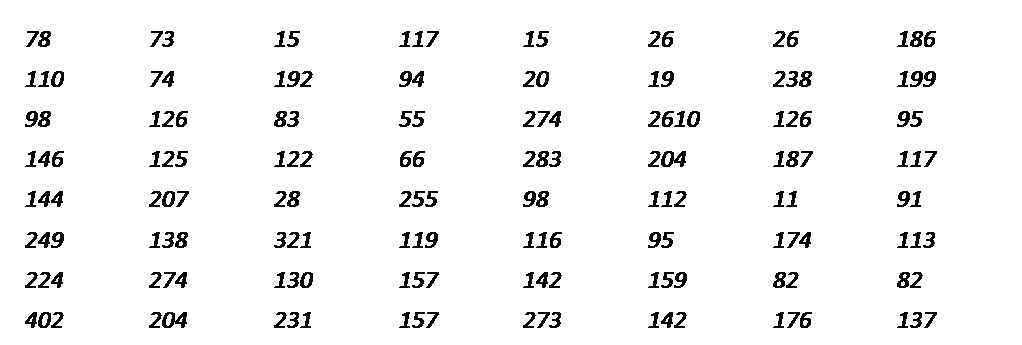
Each sequence name is followed by 2-d matrix of frequency values for 3 letter words shown above as calculated from CGR map.
Thus, one can find frequency of occurrence of any k-letter word in sequences using this tool. As shown in above example CCC has frequency of 78 in given input sequence denoted by Subtype__A
Following is the screenshot of the file for example data.

4) Outtree and Outfile – CGAT also lets users download outfile and outtree files generated by Phylip. These files contains information about trees generated in standard NEWICK format. These files are compatible with standard bioinformatics tools like TreeView , MEGA etc. to create, view , edit and customize trees.
5) NJ Tree-The tree generated by the Notung program using the Phylip Outtree input can be downloaded in PNG format.
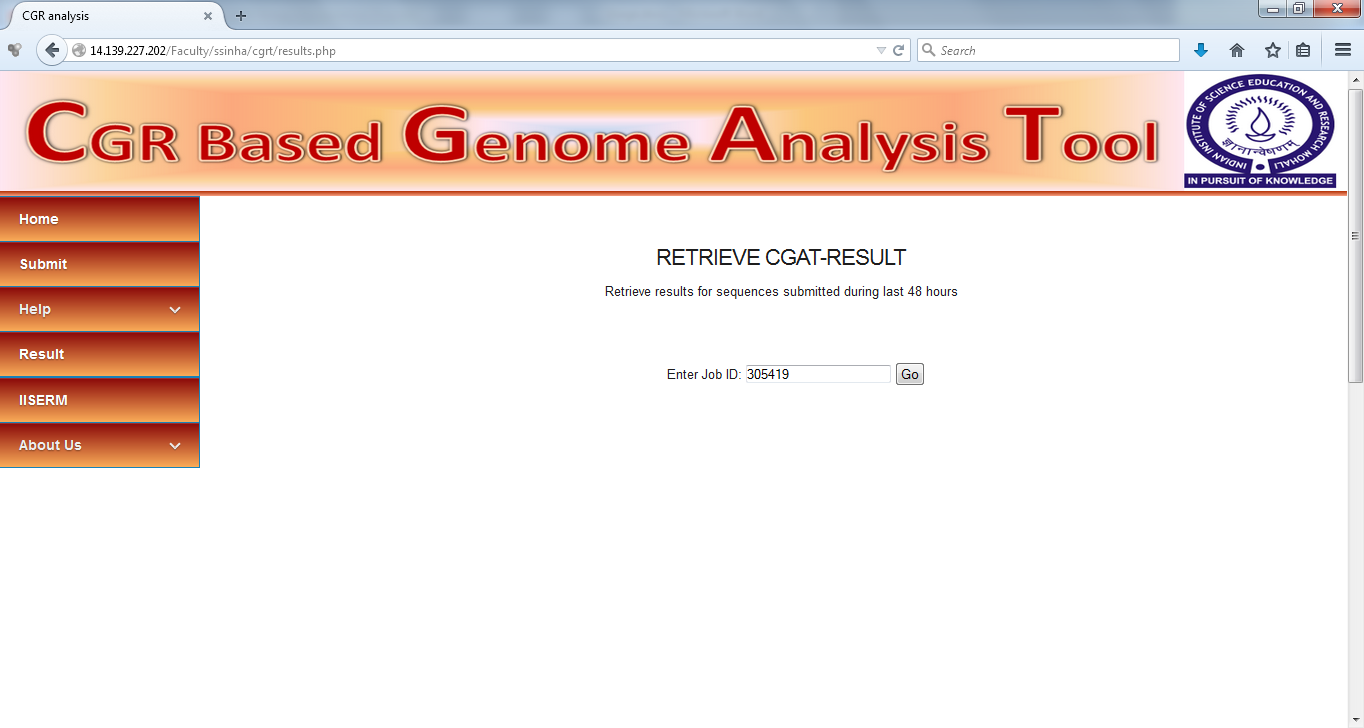
Retrieval of old results
The results of each analysis will be saved on the server for 48 hours after the analysis has been performed and these can be retrieved using the unique job id assigned at the time of analysis. For retrieving the results using job id, go the "Results" tab and submit the job id in the text box. Following is the screenshot of retrieval of the analysis performed on example data.
Contact us –
For further queries, suggestions and any technical issues regarding CGAT, send an email at thind.amarinder[at]gmail.com